

Introduction
GPX1 was the first GPX to be discovered1 and is also known as classical GPX, cytosolic GPX or GPX1. It has a wide distribution in animal cells and is present in the cystosol.1,2,3 It is reported that high levels of GPX present in different tissues such as RBC, placenta, lung, liver and kidney which parallels oxidative stress metabolism.4-6
GPX1 is capable of metabolizing hydrogen peroxides and many organic hydroperoxides species.4 However, GPX1 cannot metabolize fatty acid hydroperoxides in phospholipids, which probably occurred in cell membranes undergoing oxidative stress.5, 6
The gene for GPX1 has been cloned and sequenced in several species. It is appeared as 22-23 KDa tetrameric protein with 4 homogenous subunits, each subunit has one residue of selenocysteine.6 Sequencing and cloning experiments have shown that replacement of selenocysteine with cysteine at the active site of GPX1 results in a large diminishing in enzyme activity.7 The GPX1 gene is located on chromosome 3p21 and contains two exons.7,8,9
GPX1 is very sensitive to conditions of low selenium (Se) supply and therefore ranks low in the hierarchy of selenoproteins.10 It has been suggested that GPX1 could serve as a store for Se, which could be useful for producing another selenoprotein when levels of selenium are very limited.11,12,13 A single nucleotide polymorphism (SNP) has been described in the coding region of the GPX1 gene (T/C variation codon 198).14,15 The frequency of this variation has been reported to be altered in patients with lung cancer or breast cancer as well as in patients with the cancer of head and neck, and bladder.16,17,18,19
PCR was used to amplify specific sequences prior to analysis of the SNP in the coding region of GPX1 gene. Such a method is a suitable and useful tool for assessing these specific polymorphisms and able to deal with large numbers of human samples when little DNA is available. In this study the validation of methods to analyse single nucleotide polymorphisms in GPX1 gene have been confirmed by sequence analysis and restriction digestion analysis.
The presents research was under taken to develop a restriction fragment length polymorphism (RFLP) assay to detect and characterize GPX1 polymorphism and validate the assays by sequencing. Validation was important for analysis of the GPX1 SNP as this has not previously been analysed by RFLP among Jordanian population. The method adopted for identifying GPX1 SNP in whole blood sample for this previously mentioned population.
The contribution of this study is to explore more knowledge in different target populations and groups to specify common diseases for genetic variability with reliable and scientific tools.
Materials and Methods
Routine Analysis
Chemicals
All chemicals were purchased from Bioline or Sigma (both from UK) unless otherwise stated.
Experimental Precautions
All experimental materials such as solutions, glassware and buffers were autoclaved for a minimum of 20 minutes at 121°C to achieve optimum conditions of sterilization. Sterilization of plastic materials used in experimentation was conducted by soaking them for at least 160 minutes in DEPC-treated water followed by autoclaving.
Subject Selection
A total of three hundred and forty whole blood samples were extracted during the first six month of the year 2018 from healthy males and females with equal sex-age matched number who presented for routine laboratory analysis at Al-Hussein bin Talal University medical centre and then to identify of Single Nucleotide Polymorphisms in Selenium-Glutathione Peroxidase (GPX1) gene in the studied population. EDTA tubes were used to collect the blood samples.
Ethical Approval
All participants consented before participation to involvement in the study. Those who refused to take part in the study were excluded. Blood samples and data concerning subjects used in this study had received ethical approval from Al-Hussein bin Talal University Institutional Review Board (IRB).
Genomic DNA Isolation from Blood
Frozen whole blood (5 ml) was thawed and transferred into a polypropylene centrifuge tube and 35 ml cell lysis buffer A ( Tris-Hydrochloric acid; Conc. 10mM, and 320mM Ethylenediaminetetraacetic acid MgCl2 containing 1% Triton added after autoclaving) was added, mixed thoroughly and the mixtures were centrifuged for 10 min at 3000 rpm at 4μC. Following centrifugation, the floating fluid was removed and the particles was resuspended with 2 ml nuclear lysis buffer B (Tris-Hydrochloric; Conc. 400mM, 60mM, and 150 mM sodium chloride containing 1% sodium lauryl sulphate added after autoclaving) and this followed by the addition of 0.5 ml 5M sodium perchlorate.
The samples were mixed on a rotary mixer at room temperature for 15 minutes to fully dissolve the pellet and then incubated at 65μC for 30 min, this followed by the addition of 2.5 ml chloroform and then shaking to each tube for ten minutes. To break the emulsion (the DNA containing phase is the uppermost phase), centrifugation was done at 1500 round per minutes for 10 min. The fluid phase was then mixed gently with 2 volumes of pure (100%) cold ethanol. The DNA was seen to come out of solution as a “white cotton-wool” pellet.
Spectrophotometer Quantification of DNA
Purity of DNA samples was measured using 1μl of DNA solution diluted in a final volume of 100 μl with Diethylpyrocarbonate (DEPC)-treated water. To correct for background, a blank reading was taken using the same batch of DEPC-treated water as the one used to dilute the sample.
Extracted DNA concentration was measured by optical density 260 and 280 nm. Optical density 260 nm interpreted as 50 µg ds DNA. Whereas wavelength 280 nm corresponds to contamination of protein in the sample. The ratio of absorbance at 260 to 280 nm of 1.8 considered as sufficient DNA purity.
Agarose Gel Electrophoresis
Electrophoreses was done by 1% agarose gel using Tris acetate buffer (TAE) (10 x TAE 50mM NaOH, 10mM EDTA, and 0.4M Tris acetate pH 7.5). The appropriate weight of agarose (0.25 g) was melted in 25 ml of 1 x buffer, then cool for a few minutes, after cooling 0.68 μg /μl of ethidium bromide added and then the mixture was transferred to horizontal gel tray. To each sample (10 μl), 2 μl of 6 x loading dye was added to each sample before loading in the gel.
The gel was detected at short-wave “Ultra-Violet” light after 60 minutes electrophoresis at 100 V. The dye ethidium bromide appears as DNA bands under UV light through an orange filter using Polaroid 665 film associated with Geldoc 1000 system (Biorad).
PCR Purification
After PCR amplification, 35 μl of PCR products were purified using the PCR purification kit protocol supplied by Qiagen (Crawley, UK). This method was used to purify the PCR products from primer, nucleotides, polymerases, and salts. The purified DNA was eluted from the column by the addition of 50 μl of Elution Buffer (10mM TrisHCl pH 8.5) following manufacturer‟s instructions.
The purified PCR products were finally electrophoresed using 1% agarose gel to check the composition.20
Sequencing Reaction
After PCR amplification and purification, all the DNA samples for direct sequencing were sent to MWG Biotech (Germany). Each sequencing reaction was done by using specific primers for PCR fragments, as appropriate. Sequence of SNP sites was determined by examination of Trace files.
Statistical Analysis
Data were analysed with different statistical tests, which will be specified in each case. Throughout this work, the statistical analysis was performed using SPSS version 16 (SPSS, Inc, Chicago, IL, USA). The significance level was set at p < 0.05.
Analysis of GPX1 gene polymorphism
Experimental Design and Methods
Figure 1 shows the methods used to determine the GPX1 genotype (Pro/Leu conversion at amino acid position 198) of subjects.
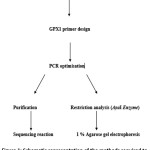 |
Figure 1: Schematic representation of the methods required to determine the GPX1 genotype (Pro/Leu 198) of subjects. Click here to View figure |
Primer Design
Primers for amplification of the appropriate part of the human GPX1 gene were designed using DNAsis (Hitachi Software). The forward primer was (5‟-GACTGCCACACGGGGATGCGTCCAT -3‟) corresponding to the nucleotides 823-848 of the human GPX1 gene sequence (Genebank, NM 000581) and the reverse primer was (5‟-TGCTGACACCCGGCACTTTATTATTAG-3‟) corresponding to nucleotides 1111-1134. The polymorphism, which results in either a Pro or Leu at amino acid position 198 is due to a C/T variation at base 911 and this position lies within the region amplified by these complementary primers (Figure 2). A computerized Genbank search was performed for these primers and they appeared to be specific, being homologous only to the human GPX1 gene.
 |
Figure 2: The section of the human GPX1 Gene sequence around the polymorphism at amino acid position 198. Click here to View figure |
The underlined section shows primers positions corresponding to the appropriate part of the human GPX1 gene and the polymorphism where a C/T variation (base 911) results in either a Pro or Leu at amino acid position 198.
PCR Conditions
In order to amplify genomic DNA, PCR wa performed using a master mix containing 5 μl of deoxynucleotide triphosphate (dNTP; Bioline, stock at 25 mM), 1μM of each primers was added to a final concentration (MWG-BIOTECH), 5 μl of 10x Buffer (Bioline), 2.5 μl MgCl2 (Bioline; stock at 50 mM), 0.4 μl Taq polymerase enzyme mix (5 units; Bioline) and DEPC- treated water to complete the final reaction volume (49 μl). This was prepared in a room dedicated to PCR where no nucleic acids were handled. Then back in the main laboratory, 0.6μg of the DNA described above was added to the master mix and then PCR performed by using a Sanyo machine.
A tube containing the master mix with no DNA added (no template control) was carried out for each PCR to check the purity of the master mix used in the preparation of the PCR reaction.
The PCR reactions were performed in triplicate in 50 μl volume, the products obtained were pooled and purified using the PCR purification protocol kit supplied by Qiagen (Catalog No./ID:28106). The purified DNA was eluted from the column by the addition of 50 μl DEPC-treated water. Agarose gel was used to confirm the quality of all PCR products.
Restriction Analysis
PCR product (10 μl) separated by using 1 % agarose gel, and bands at the expected size were visualized. The PCR products, unpurified, were then subjected to RFLP analysis. The appropriate volume (3μl) of the reaction was digested with ApaI enzyme (Promega; Southampton, UK). To carry out the restriction analysis, 3μl of PCR product was added to 2μl enzyme (5 U), 2 μl restriction enzyme 10 x buffer A (Promega; Southampton, UK), 2μl BSA (1 mg/ml, Promega; Southampton, UK) to enhance the activity of the ApaI restriction enzyme and DEPC- treated water to complete the final reaction volume (20 μl) . Samples were incubated for approximately one hour at 37μC. The samples were then analysed by agarose gel electrophoresis.
Results
Validation of PCR of the GPX1 gene
PCR of human GPX1
Figure 3 shows the results of gel analysis of the PCR product using primers aligned to amplify relevant parts of GPX1 gene. The PCR products showed a band at the expected size of approximately 311 bp with no sign of genomic contamination. To confirm that the amplification product is corresponded to the appropriate part of the GPX1 gene, a restriction analysis using ApaI enzyme (the reaction was incubated for 1 hr at 37μC) and sequencing of the PCR fragment using the PCR primers as sequencing primers, were performed (as described in section 2.1.7). Restriction analysis and sequence analyses confirmed the identity of the amplification product as the correct part of the GPX1 gene, indicating the specificity of the primers designed for the GPX1 gene and showing that the 311 bp product is corresponded to region 823-1134 of the human GPX1 gene.
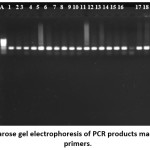 |
Figure 3: Agarose gel electrophoresis of PCR products made using GPX1 primers. Click here to View figure |
Lane A is a hyperladder, Lane B is a negative control, lanes 1-19 represent PCR products using genomic DNA from different individuals. Samples were analysed using a 1% agarose gel.
Restriction fragment length polymorphism analysis of part of the human GPX1
To detect the three possible forms (genotypes) of the human GPX1 polymorphism in codon 198, RFLP analysis was performed using the restriction enzyme ApaI as described in section 2.2.4. Sequence analysis shows ApaI site at the site of SNP, this could potentially be used in RFLP analysis.
When the codon encoding proline, (CCC), is present, two DNA fragments of 223 and 88bp are produced (Figure 4; e.g. lanes 1, 3, 4, and 9). In contrast the Leu/Leu (CTC) genotype is predicted not to be cut by ApaI resulting in 311 bp (Figure 4; e.g. lane 10). The Pro/Leu (heterozygote) genotype produces 3 fragments of 88, 223, and 311 bp respectively (Figure 4; e.g. lanes 2, 5, 6, 7 and 8). Subjects represented in the gel were assigned to one of three possible genotypes: two homozygotic, the Leu/Leu (CTC) genotype, and the Pro/Pro (CCC) genotype and the Pro/Leu (heterozygote) as shown in Table 1.
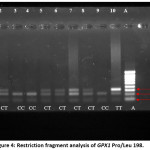 |
Figure 4: Restriction fragment analysis of GPX1 Pro/Leu 198. Click here to View figure |
The figure shows the results of restriction analysis of DNA from 10 individuals. Lane A is a hyperladder marker; lanes 1-10 contain the PCR product from separate people after digestion with ApaI enzyme. Restriction analysis yields one (lane 10), two (lane 1, 3, 4, 9) or three (lanes 2, 5, 6, 7, 8) DNA fragments according to genotype.
Sequencing analysis of PCR reactions of human GPX1
To identify the SNP Pro/Leu 198 in the human GPX1 gene, and in addition to check that RFLP gave appropriate results, sequencing of products of PCR of the GPX1 fragment was performed as described in section 2.1.7.
Products of the 10 reactions corresponding to those analysed by RFLP (Figure 4) were purified and sent for sequencing by MWG. Figure 5 shows the result of sequence analysis for three samples. The trace file of the sequence obtained from these three subjects was examined and aligned with the published sequence of the human GPX1 gene sequence. The sequences corresponded to the part of the human GPX1 gene containing the SNP Pro/Leu 198, which is due to C/T variation at base 911 (approximately at position 60 in the Figure 5). This polymorphism was present in three forms; two homozygotes, the Leu/Leu (CTC) genotype, and the Pro/Pro (CCC) genotype and a heterozygote form the Pro/Leu.
Subjects sequenced by MWG were assigned to one of the three possible genotypes as shown in Table 1 and the genotype derived by sequencing corresponded exactly with that found by restriction analysis.
 |
Figure 5: Trace files of the GPX1 sequences obtained from PCR products derived from human DNA. Click here to View figure |
Purified PCR prodcts were sent to MWG for sequenceanalysis. The SNP Pro/Leu 198 in the human GPX1 gene (approximately at position 60 in the figure). The green line shows the Pro/Pro (CCC) genotype, the red line shows the Pro/Leu (heterozygote) genotype and the yellow line shows the Leu/Leu (CTC) genotype.
Table 1: Comparison of GPX1 genotype results from RFLP analysis by ApaI enzyme and sequencing analysis.
| Subject | GPX1 genotype | |
| RFLP | Sequencing | |
| 1 | CC | Pro/Pro |
| 2 | CT | Pro/Leu |
| 3 | CC | Pro/Pro |
| 4 | CC | Pro/Pro |
| 5 | CT | Pro/Leu |
| 6 | CT | Pro/Leu |
| 7 | CT | Pro/Leu |
| 8 | CT | Pro/Leu |
| 9 | CC | Pro/Pro |
| 10 | TT | Leu/Leu |
Discussion
Recently a SNP in the coding region of GPX1 has been identified; this is located at codon 198 in the GPX1 gene at base position 911 and the variants detected by RFLP (Moscow et al., 1994; Forsberg et al. 2000). 14, 15
The first aim of this study was to develop an RFLP method for analyses of the GPX1 and to validate the assay by sequencing. For analysis of the GPX1 SNP at the specified base position, amplification by polymerase chain reaction (PCR) resulting in a product of 311 bp (Figure 3) and RFLP with ApaI enzyme allowed identification of Pro/Pro, Pro/Leu and Leu/Leu genotypes (Figure 4). The Pro/Pro is recognised by ApaI resulting in the cleavage of the DNA fragment into 2 fragments of 88 and 223bp respectively. The rare Leu/Leu is not recognised by the enzyme resulting in an uncut product. The Pro/Leu (heterozygote) genotype produces 3 fragments of 88, 223, and 311 bp respectively. The results from the RFLP of all of the samples analysed in this validation exercise were confirmed by direct sequencing (MWG, Ebersberg, Germany) with 100% agreement (Figure 5).
These results were compared with several studies that were looking at the effects of The GPX1 SNP (Pro/Leu, codon 198) in lung cancer patients and breast cancer patients, as well as in patients with cancer of the head and neck respectively, and reported the frequency of this polymorphism in cancer-free people. 16,17,18,21 All these studies have indicated that the RFLP method was reliable and valid as a tool for identifying these polymorphisms with a high degree of specificity and sensitivity.
The results of the experiment to develop a method for GPX1 SNP analysis showed that the PCR products of the fragment of the GPX1 gene corresponds to the published sequence of the human GPX1 gene between nucleotides 823 and 1134, that restriction analysis with ApaI was able to distinguish between T/C genotypes and that the restriction analysis gives a rapid method for identifying the different polymorphisms with a high degree of specificity and sensitivity, which will be suitable for analysis of the large number of samples required for future studies.
Conclusion
The RFLP assay, which had been developed and then validated by different strategies including direct sequencing, was chosen in this study as means of identifying the polymorphisms in the target gene, GPX1. This study demonstrated that the RFLP method was rapid, and reliable and valid as a tool for identifying the different polymorphisms with high degree of specificity. Other approaches such as these based on real-time PCR technology might be applied for much larger populations.
Acknowledgements
Authors are grateful to Dr. Amin N. Olaimat for valuable technical assistance.
Funding
This work was funded from the deanship of scientific research at Al-Hussein bin Talal University, Ma’an, Jordan.
Conflict of Interest
The authors declare no potential conflict of interests.
References
- Rotruck J.T., Pope A. L., Ganther H.E., Swanson A.B., Hafeman D.G., Hoekstra W.G. Selenium: Biochemical Role as a Component of Glutathione Geroxidase. Science. 1973; 179: 588-590.
- Brigelius-Flohe R. Tissue-Specific Functions of Individual Glutathione Peroxidases. Free Radicals in Biology and Medicine. 1999; 27: 951-965.
- Evenson J.K., Wheeler A.D., Blake S.M., Sunde R.A. Selenoprotein mRNA is expressed in blood at levels comparable to major tissues in rats. Journal of Nutrition. 2004; 134: 2640-2645.
- Sunde R. The Biochemistry of Selenoproteins. J Am Oil Chem Soc. 1984; 61: 1891-1900.
- Grossman A.A. Wendel. Non-Reactivity of the Selenoenzyme Glutathione Peroxidase with Enzymatically Hydroperoxidized Phospholipids. European Journal of Biochemistry.1983; 135: 549-552.
- Forstrom J. W., Zakowski J. J., Tappel A. L. Identification of the Catalytic Site of Rat liver Glutathione Peroxidase as Selenocysteine. Biochemistry.1978; 17: 2639-2644.
- Rocher C., Lalanne J.L., Chaudiere J. Purification and Properities of Recombinant Sulfur Analog of Murine Selenium-Glutathione Peroxidase. Eur. J.Biochem.1992; 205: 955- 960.
- Kiss C. Li J., Gizatullin R.Z., Kashuba V.I., Lushnkiova T., Protopopov A.I., Kelve M., Kiss H., Kholodnyuk I.D., Imreh S., Klein G., Zabarovsky E.R., Cytogenet. Cell Genet. 1997; 79: 228-230.
- Ishida K., Morino T., Takagi K., Sukenaga Y. Nucleic Acids Res.1987; 15:p 10051.
- Brigelius-Flohe R. Tissue-Specific Functions of Individual Glutathione Peroxidases. Free Radicals in Biology and Medicine.1999; 27: 951-965.
- Burk R.F., Gregory P.E. Some Characteristics of 75Se-P, a Selenoprotein found in Rat liver and Plasma, and Comparison of it with Selenoglutathione Peroxidase. Archives of Biochemistry and Biophysics.1982; 213: 73-80.
- Burk R.F., Hill K.E. Regulation of selenoproteins. Annual Review of Nutrition.1993; 13: 65-81.
- Sunde R.A. Regulation of Selenoprotein Gene Expression, ed. Hatfield DL. In: Selenium: its Molecular Biology and Role in Human Health. 2001; 81-96. Boston, Kluwer Academic Publishers.
- Moscow J.A., Schmidt L., Ingram D.T., Gnarra J., Johnson B., Cowan K.H. Loss of Heterozygosity of the Human Cytosolic Glutathione Peroxidase I gene in Lung Cancer. Carcinogenesis. 1994; 15: 2769-2773.
- Forsberg L., de Faire U., Marklund S.L., Andersson P.M., Stegmayr B., Morgenstern R. Phenotype Determination of a Common Pro-Leu Polymorphism in Human Glutathione Peroxidase 1. Blood Cells Molecules and Disease. 2000; 26: 423-426.
- Ratnasinghe D., Tangrea J.A., Andersen M.R., Barrett M.J., Virtamo J., Taylor P.R., Albanes D. Glutathione Peroxidase Codon 198 Polymorphism Variant Increases Lung Cancer Risk. Cancer Research.2000; 60: 6381-6383.
- Hu Y.J., Diamond A. M. Role of Glutathione Peroxidase 1 in Breast Cancer: Loss of Heterozygosity and Allelic Differences in the Response to Selenium. Cancer Research. 2003; 63:3347-3351.
- Hu Y.J., Dolan M.E., Bae R., Yee H., Roy M., Glickman R., Kiremidjian-Schumacher L., Diamond A.M. Allelic Loss at the GPx-1 Locus in Cancer of the Head and Neck. Biological Trace Element Research. 2004; 101:97-106.
- Ichimura Y., Habuchi T., Tsuchiya N., Wang L., Oyama C., Sato K., Nishiyama H., Ogawa O., Kato T. Increased Risk of Bladder Cancer Associated with a Glutathione Peroxidase 1 codon 198 variant. Journal of Urology. 2004; 172: 728-732.
- Leonard J.T., Grace M.B., Buzard G.S., Mullen M.J., Barbagallo C.B. Preparation of PCR Products for DNA Sequencing. BioTechniques. 1998; 24:314-317.
- Cox D.G., Hankinson S.E., Kraft P., Hunter D.J. No Association between GPX1 Pro198Leu and Breast Cancer risk. Cancer Epidemiology Biomarkers and Prevention. 2004; 13: 1821-1822

This work is licensed under a Creative Commons Attribution 4.0 International License.






